Meta AI introduces UMA (atomic general model): atomic general model family
Density Function Theory (DFT) is the foundation of modern computational chemistry and materials science. However, its high computational cost severely limits its use. The interatomic potential (MLIP) of machine learning has the potential to closely approximate DFT accuracy while significantly improving performance, reducing computation time from hour to less than one second, while O(n) is compared to O(n³) scaling. However, training MLIP across different chemistry tasks remains an open challenge, as traditional methods rely on smaller problem-specific datasets rather than using scaling advantages that have made significant advances in language and visual models.
Existing attempts to address these challenges focus on developing general-purpose MLIPs trained on larger datasets, while datasets such as Alexandria and Omat24 can improve performance on the MATBENCH-DISCOVERY rankings. Additionally, researchers have explored extended relationships to understand the relationship between computation, data and model size, drawing inspiration from the empirical scaling laws in LLMS that facilitate training more tokens and have larger models to improve predictable performance. These scaling relationships help determine the optimal resource allocation between the dataset and model size. However, their application on MLIP is still limited compared to the transformative impacts seen in language modeling.

Researchers from Fair from Meta and Carnegie Mellon University have proposed a family of atomic universal models (UMA) designed to test the limits of accuracy, speed and generalization across chemical models in chemistry and materials science. Furthermore, to address these challenges, they developed empirical scaling laws related to calculations, data, and model sizes to determine the optimal model size and training strategies. This helps overcome the challenges of balancing accuracy and efficiency, due to an unprecedented data set of about 500 million atomic systems. Furthermore, UMA’s accuracy and inference speed are similar or better than professional models on a wide range of material, molecular and catalytic benchmarks without fine-tuning for specific tasks.
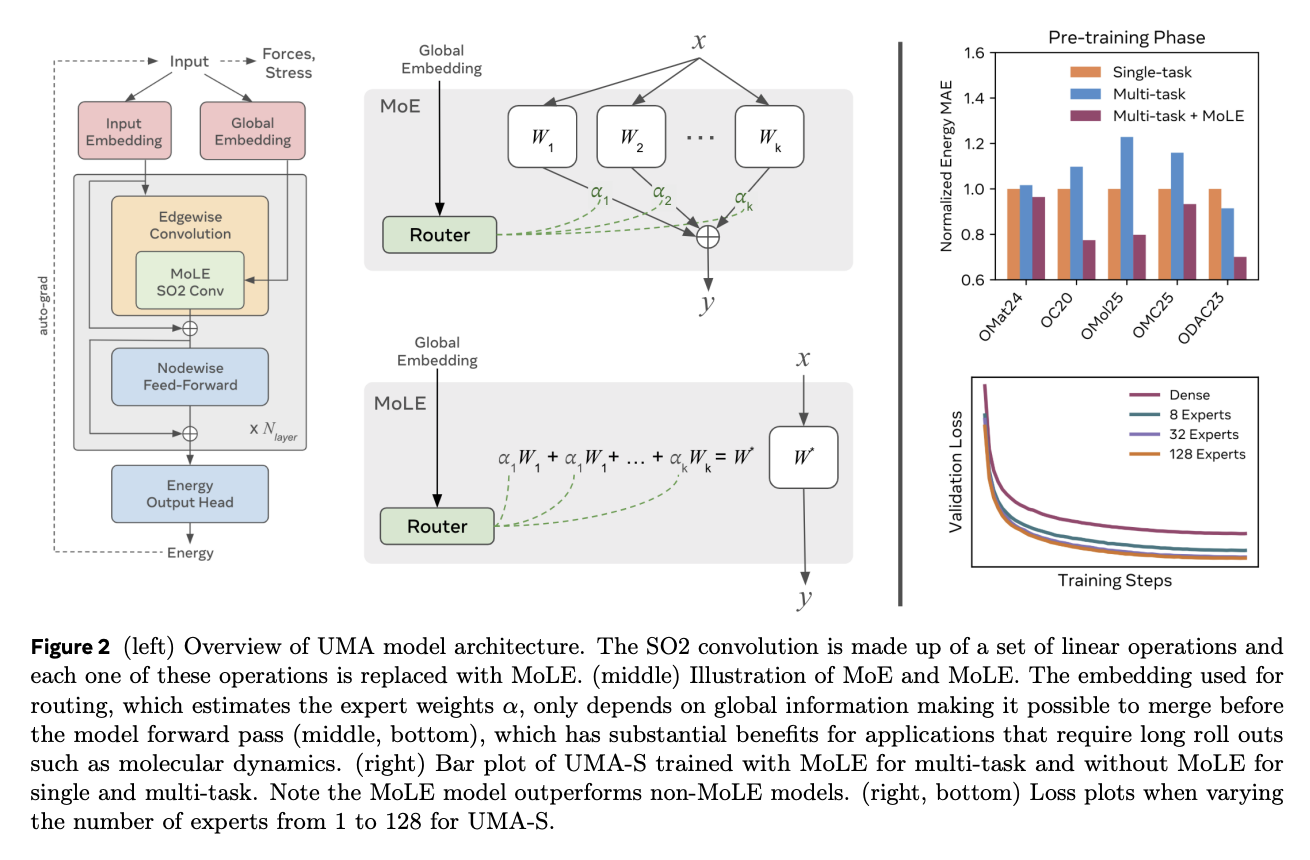
The UMA architecture is built on Esen (Esen), a fuzzy graphical neural network with critical modifications that enable efficient scaling and process other inputs, including total charges for simulation, spin and DFT settings. It also incorporates a new embedded approach that allows UMA models to integrate charge, rotation and DFT-related tasks. Each of these inputs generates an embedding of the same size as the spherical channel used. The training follows a two-stage approach: the first stage directly predicts forces for faster training, and the second stage will eliminate force heads and fine-tune the model to predict the use of automatic levels of force and pressure, thus ensuring energy protection and smooth potential energy landscape.
The results show that the UMA model exhibits logarithmic linear scaling behavior over the tested flop range. This suggests that larger model capacity is needed to fit the UMA dataset, these scaling relationships are used to select accurate model sizes and to show the advantages of molar-ratio-intensive architectures. In multitasking training, the migration from 1 expert to 8 experts, with the smaller gains of 32 experts and the improvements of 128 experts were negligible, and significant improvements were observed. Furthermore, despite the large parameter count, the UMA model still exhibits extraordinary inference efficiency and the UMA-S is able to simulate 1000 atoms in 16 steps per second and fit system sizes up to 100,000 atoms in memory on a single 80GB GPU.
In summary, the researchers introduced a family of atomic universal models (UMA) that exhibits strong properties across a wide range of benchmarks, including materials, molecules, catalysts, molecular crystals and metal organic frameworks. It achieves new and latest results on established benchmarks such as Adsorbml and Matbench Discovery. However, it cannot handle remote interactions due to the standard 6Å cutoff distance. Additionally, it uses a separate embedded type for discrete charge or spin values, which will generalize to limit the invisible charge or rotation. Future research aims to move towards universal MLIP and unlock new possibilities in atomic simulations, while highlighting the need for more challenging benchmarks to drive future progress.

Sajjad Ansari is a final year undergraduate student from IIT Kharagpur. As a technology enthusiast, he delves into the practical application of AI, focusing on understanding AI technology and its real-world impact. He aims to express complex AI concepts in a clear and easy way.


